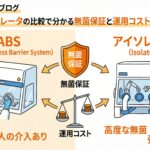
再生医療製品の製造において、無菌性の保証は製品の品質、ひいては患者様の安全に直結する最重要課題です。特に、アイソレータやRABS(制限アクセスバリアシステム)を使用する際、作業者と無菌エリアを隔てる「グローブ」は、バリアシステムの中で最も脆弱な部分といわれています。
目に見えない微細なピンホールひとつが、重大な汚染事故を引き起こすリスクを孕んでいるのです。そのため、グローブの完全性を科学的に証明する「グローブリーク試験」の適切な運用は、製造現場における品質保証の要といえるでしょう。
この記事では、PIC/S GMP Annex 1の改訂など最新の規制動向を踏まえつつ、グローブリーク試験の具体的な手法や、現場の負担を軽減する試験機の選定ポイントについて詳しく解説します。確実な無菌管理体制の構築にお役立てください。
グローブリーク試験とは|無菌操作における重要性と目的

グローブリーク試験とは、アイソレータやRABSに設置されたグローブにピンホールや破損がないかを確認し、バリアとしての完全性が保たれているかを検証する検査のことです。
無菌操作の信頼性を担保するために、なぜこの試験がこれほどまでに重要視されているのか、その背景と目的について掘り下げていきましょう。
アイソレータ・RABSにおけるグローブの完全性確保
アイソレータやRABSにおいて、グローブは唯一、柔軟性のある素材で構成され、かつ頻繁に物理的なストレスがかかるパーツです。ステンレスやガラスで構成された強固な筐体とは異なり、グローブは操作のたびに伸縮を繰り返すため、経年劣化や鋭利な器具との接触による破損リスクが常に存在します。
グローブの完全性を確保することは、すなわち無菌エリア(グレードA)の環境を維持することと同義です。定期的なグローブリーク試験によって、目視では発見できない微細な欠陥を検出し、バリア機能が正常に働いていることを客観的なデータとして証明する必要があります。
再生医療等の製造現場でグローブリーク試験が必須とされる理由
再生医療等製品の多くは、最終的な滅菌工程(ターミナルステリライゼーション)を適用できないため、製造プロセスの全工程において無菌性を維持する「無菌操作法」が必須となります。
もし製造中に微生物汚染が発生すれば、貴重な細胞製品を廃棄せざるを得ないだけでなく、患者様への供給遅延や健康被害といった深刻な事態を招きかねません。
グローブリーク試験は、このような汚染リスクを未然に防ぎ、製造プロセスの堅牢性を証明するための必須要件として、製造管理および品質管理の基準(GMP/GCTP)の中でも特に重要視されています。
ピンホールによる無菌性保証リスクと汚染事故の防止
グローブに生じたピンホールは、たとえ肉眼では確認できないほどの微細な穴であっても、微生物にとっては十分な侵入経路となり得ます。アイソレータ内部が陽圧に保たれていたとしても、操作時の圧力変動(ポンピング作用)によって、外部の空気が内部に吸い込まれ、汚染を引き起こす可能性があるのです。
適切な感度を持つ試験機を用いて定期的に検査を行うことは、こうした「見えないリスク」を可視化し、排除するための唯一の手段です。汚染事故を未然に防ぐことは、コスト削減だけでなく、企業の信頼性を守ることにもつながります。
グローブリーク試験における規制要件とガイドライン
医薬品や再生医療等製品の製造において、グローブの管理は各国の規制当局によって厳格に定められています。
ここでは、特に影響力の大きいPIC/S GMP Annex 1の改訂内容を中心に、日本薬局方やFDAガイドラインにおけるグローブリーク試験の位置づけについて解説します。
PIC/S GMP Annex 1改訂に伴う管理基準の厳格化
無菌医薬品製造の国際的なガイドラインであるPIC/S GMP Annex 1の改訂(2022年発行)において、グローブの管理基準は大幅に強化されました。
具体的には、アイソレータやRABSのグローブに対して、物理的および機械的な完全性試験を実施することが明確に求められています。
注目すべきは、試験の頻度に関する記述です。「使用前および使用後」の実施が推奨されており、さらにキャンペーン生産などの連続運転時における試験間隔についても、リスク評価に基づいた設定が必要とされています。これにより、従来の管理手法の見直しを迫られる現場も少なくありません。
日本薬局方(JP)およびISO 14644-7における記載
日本薬局方(JP)においても、無菌医薬品製造区域の管理に関する参考情報として、バリアシステムの完全性確保について言及されています。
また、ISO 14644-7(分離装置)では、分離装置のリークテストに関する具体的な手法や基準が示されており、グローブシステムもその対象に含まれます。
これらの規格では、単に試験を行うだけでなく、試験方法が科学的妥当性を持ち、検出限界が明確であることを求めています。つまり、使用する試験機がJISやISOなどの規格に準拠した測定原理に基づいていることが、バリデーションの観点からも重要となるのです。
FDAガイドラインにおけるグローブ管理の指針
日本の無菌操作法指針(FDAの2004年ガイドライン等を参考に策定)において、グローブは無菌環境を維持するための「重要なバリア」として位置づけられています。指針では、グローブの破損が製品汚染のリスクにつながる可能性があることから、適切な管理体制の構築が求められているのです。
具体的には、使用前後の目視確認に加え、定期的なグローブリーク試験を実施することが望ましいとされています。目視検査だけでは微細なピンホール等の発見が難しい場合もあるため、より感度の高い物理的な試験方法を併用することが推奨されています。
また、日々の運用においては、試験の実施記録を適切に管理するだけでなく、万が一リークが検出された際に備え、製品への影響評価(インパクトアセスメント)を行う手順を定めておくことも重要でしょう。確実な記録と手順の整備は、製品の品質保証において欠かせない要素となります。
グローブリーク試験の主な手法と測定原理
グローブリーク試験にはいくつかの手法が存在しますが、JIS規格(JIS Z4820)等に基づき、目的に応じた適切な方法を選定することが大切です。
主に用いられるのは、グローブボックスの気密試験で標準とされる「大気圧比較法」や「漏れなし容器法」、また製薬用アイソレータで活用される「陽圧減衰法」などでしょう。
ここでは、これら主要な手法の原理と特徴、そして実務的な視点から見た違いについて解説します。
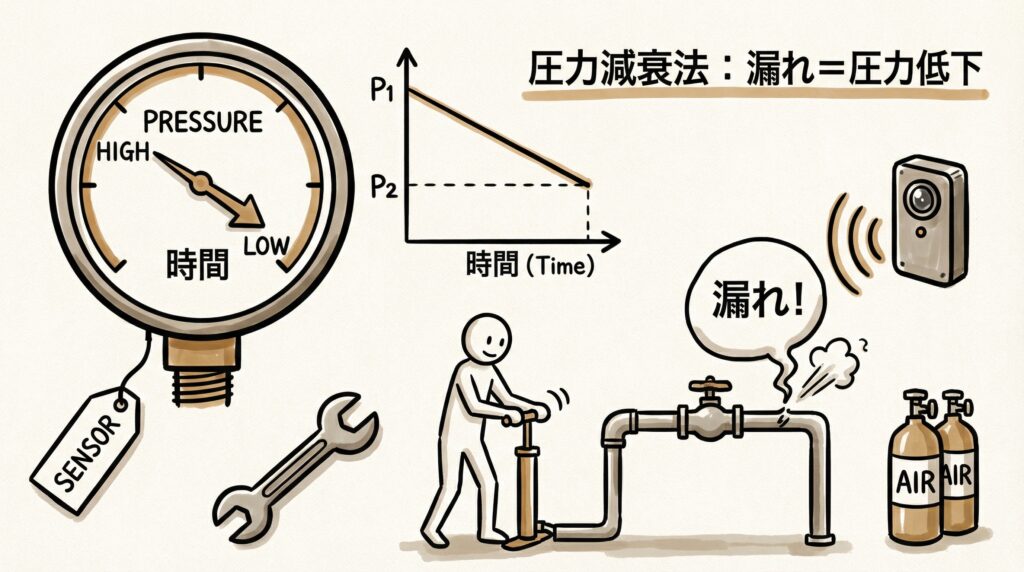
圧力減衰法(JIS T 9116準拠)の仕組み
圧力減衰法(Pressure Decay Method)は、JIS T 9116などの規格にも準拠した、信頼性の高い試験方法です。
具体的な仕組みは以下の通りです。
- 専用のテストポートを用いてグローブ開口部を密閉する。
- グローブ内部に清浄な空気を送り込み、規定の圧力まで加圧して膨らませる。
- 空気の供給を止め、一定時間(安定化時間および計測時間)圧力を保持する。
- この間の圧力低下量を測定し、あらかじめ設定した閾値を超えて圧力が下がった場合を「リークあり(不合格)」と判定する。
この方法は、グローブを破壊することなく(非破壊検査)、客観的な数値データとして結果を得られるのが特徴です。
圧力減衰法のメリットとデメリット
圧力減衰法は多くの現場で採用されていますが、メリットとデメリットの両面を理解しておくことが大切です。
- メリット
- 自動化が可能で、測定者による個人差が出にくい。
- 数値データとして記録が残るため、データインテグリティ(DI)の観点で優れている。
- グローブを汚染することなく、設置した状態で試験(インサイチュ試験)が可能。
- デメリット
- 温度変化の影響を受けやすく、空調の風が当たると圧力が変動し誤検知の原因になることがある。
- グローブの材質(ゴムの弾性)による影響(クリープ現象)を考慮する必要がある。
- 試験にある程度の時間(数分〜十数分)を要する。
その他の試験法(アンモニアリーク法・水没法)との比較
再生医療の製造現場では、アイソレーターの気密性を担保するグローブリーク試験に加え、製品そのものの無菌性を確認する試験法の選定も極めて重要です。ここでは、従来の公定法と、近年再生医療分野で導入が進む迅速法の特徴を比較してみましょう。
| 試験方法 | 概要 | 特徴 |
|---|---|---|
| 直接法・メンブランフィルター法 | 培地を用いて微生物を増殖・検出 | 公定法。信頼性は高いが、結果判定に14日間程度を要するため、出荷判定のボトルネックになりやすい。 |
| 生物発光法 | 微生物が持つATPなどを測定して検出 | 迅速な判定が可能。ただし、感度やバックグラウンドの影響を考慮した運用が必要。 |
| 固相サイトメトリー法 | 蛍光染色した菌をレーザーで検出 | 迅速法の代表格。培養不要で短時間の測定ができ、有効期限の短い再生医療製品に適している。 |
製造現場における品質管理としては、グローブリーク試験(圧力減衰法等)によってハード面の無菌環境を維持しつつ、製品特性に合わせて核酸増幅法や固相サイトメトリー法などの迅速試験を適切に取り入れることが、最も合理的かつ現実的なアプローチといえるでしょう。
適切な試験頻度と運用のタイミング
「いつ、どのタイミングで試験を行うべきか」は、運用の効率とリスク管理のバランスを左右する重要なテーマです。
規制要件を満たしつつ、製造スケジュールを圧迫しない最適な試験頻度と、異常時の対応フローについて解説します。
作業前・作業後の試験実施の重要性
PIC/S GMP Annex 1の改訂により、クリーンルームのクラス確認や微生物モニタリングにおいて、「作業前(at-rest)」の状態だけでなく、「作業中(in-operation)・作業後」の実施も強く求められるようになりました。
- 作業前の確認(At-rest): 製造開始前に微粒子や微生物のモニタリングを行い、規定の環境清浄度が維持されているかを保証するために不可欠です。この段階で、バリアシステムの完全性を裏付けるグローブリーク試験などの実施計画も、汚染管理戦略(CCS)において重要な要素となります。
- 作業中・後の確認(In-operation): 製造プロセスを通じて無菌環境が損なわれなかったことを証明するために行います。もしモニタリングで異常が認められた場合は、リスク評価に基づいた対応が必要となるため、製品の品質を担保する上で極めて重要な意味を持つでしょう。
毎回の作業前後に厳密な確認を組み込むことは現場の負担になりますが、リスクベースのアプローチを取り入れ、手順を標準化していくことが大切です。
キャンペーン生産時における試験間隔の考え方
数日間にわたって連続生産を行う「キャンペーン生産」の場合、毎回の作業後に試験を行うことが難しいケースもあります。
このような場合は、リスクアセスメントに基づき、科学的妥当性のある試験間隔を設定する必要があります。
例えば、「毎日の作業終了時に実施する」「特定の重要工程の前後で実施する」といったルールを定めます。ただし、間隔を空ければ空けるほど、万が一リークが発覚した際に廃棄対象となる製品(ロット)の範囲が広がるリスクがあることを考慮しなければなりません。
生産効率とリスクのトレードオフを慎重に検討しましょう。
万が一リークが検出された場合の逸脱管理と処置フロー
試験で「不合格(Fail)」となった場合の対応フローをあらかじめ定めておくことは必須です。
単に再試験をして合格すれば良い、というものではありません。
- 原因調査: ポートへの装着ミスか、グローブ自体の破損かを確認。
- 製品への影響評価: 破損箇所、破損の大きさ、作業内容、環境モニタリングの結果などを総合的に評価し、当該ロットの製品品質への影響を判断する。
- 是正措置・予防措置(CAPA): グローブの交換、手順の見直しなどを行い、再発防止を図る。
これらのプロセスを逸脱管理手順として文書化し、適切に運用することが求められます。
失敗しないグローブリークテスター(試験機)の選定ポイント
の選定ポイント-1024x572.jpg)
グローブリークテスター(試験機)は、一度導入すると長く使用する機器です。現場の作業性やコンプライアンス対応を左右するため、機種選定は慎重に行う必要があります。
失敗しないための具体的なチェックポイントを5つご紹介します。
ポート形状とカフリングへの適合性確認
最も基本的なポイントですが、自社のアイソレータのグローブポート形状に、テスターが確実にフィットするかどうかを確認しましょう。
ポートの直径やフランジの形状はメーカーによって異なります。
多くのテスターは専用のアタッチメントやカフリングを使用しますが、装着が難しかったり、密閉性が不安定だったりすると、試験のたびにエラー(偽陽性)が発生し、業務効率が著しく低下します。
デモ機を借りて、実際のポートでスムーズに装着・密閉できるかを必ずテストすることをおすすめします。
ワイヤレス方式と有線方式の操作性比較
グローブリーク試験に使用するテスターには、制御盤とケーブルやエアチューブで接続された「有線方式」と、バッテリー駆動で動作する「ワイヤレス方式」のモデルが存在します。
- ワイヤレス方式: ケーブルの取り回しに気を遣う必要がなく、複数のポートを移動して試験を行う際にスムーズに作業できるでしょう。配線が床や機器周辺を這わないため、清掃時の妨げになりにくい点もメリットです。一方で、バッテリーの充電管理といった運用上の手間は考慮しなければなりません。
- 有線方式: 電源供給が安定しており、バッテリー切れの心配がないため、長時間の連続使用でも安心感があります。ただし、接続ケーブルが作業者の動線や周辺機器と干渉しないよう、配置には工夫が求められます。
最近では、測定データを無線通信で転送し、データ管理を効率化できる機能を備えたモデルも登場しています。施設のレイアウトや日々の運用フローを考慮し、最適なタイプを検討してみてはいかがでしょうか。
測定時間の短縮と生産効率への影響
アイソレータには通常、複数のグローブ(例えば4本〜10本以上)が設置されています。1本あたりの測定時間が長ければ、全体の準備時間は膨大なものになります。
例えば、1本の測定に15分かかる機種と、5分で完了する機種では、トータルの拘束時間が大きく異なります。
「高速測定モード」を搭載している機種や、複数のグローブを同時に並行して測定できるシステムを選定することは、生産効率を高める上で非常に有効な投資となります。
Data Integrity(データインテグリティ)対応と記録管理機能
医薬品製造の現場では、データの完全性(Data Integrity)が厳しく問われます。
試験結果が紙のレシート出力だけでは、データの紛失や改ざんのリスクを指摘される可能性があります。
- ユーザー権限管理(誰が試験したか)
- 監査証跡(いつ、どのような操作をしたか)
- 電子記録の保存とバックアップ
これらの機能が搭載された、21 CFR Part 11対応のソフトウェアを持つ機種を選ぶことが、将来的な査察対応を見据えた際の安心材料となります。
現場での校正(キャリブレーション)のしやすさ
測定機器である以上、定期的な校正(キャリブレーション)は必須です。しかし、メーカーに機器を送り返して校正を行う期間中、試験ができなくなるのは困ります。
- 現場でユーザー自身が日常点検(簡易チェック)できる機能があるか
- 外部の参照圧力計を用いて、現場で比較校正が可能か
- メーカーの出張校正サービスが充実しているか
これらを確認し、運用のダウンタイムを最小限に抑えられる体制を整えておくことが大切です。
グローブリーク試験のバリデーションと日常管理
試験機を購入して終わりではありません。導入時の適格性評価(バリデーション)から日常のメンテナンスまで、一貫した管理体制を構築することで初めて、試験結果の信頼性が保証されます。
運用のフェーズごとにやるべきことを整理しましょう。
導入時のバリデーション(IQ/OQ/PQ)の進め方
新しい試験機を導入する際は、IQ(据付時適格性確認)、OQ(運転時適格性確認)、PQ(性能適格性確認)を実施します。
- IQ: 機器が正しく設置され、仕様通りであることを確認。
- OQ: 正常に動作し、設定した機能が働くことを確認。
- PQ: 実際のグローブを用いて、意図したピンホール(標準穴)を確実に検知できるかを検証。
特にPQでは、自社の運用条件(グローブの種類、試験圧力、時間)において、検出したいピンホールサイズ(例:100μm)を検知できるパラメータ(レシピ)を確立することが最重要です。
定期的な点検とグローブの交換基準
グローブリーク試験はあくまで「今の状態」を確認するものです。グローブの劣化傾向を把握し、予防的に交換する基準を設けることも重要です。
- 定期交換: 「半年ごと」「〇〇バッチごと」など、使用頻度に応じた交換サイクルを定めます。
- 日常点検: 試験機による検査に加え、使用前には必ず作業者による目視確認(傷、変色、べたつき等)を行います。
機械的な試験と人間の目によるチェックを組み合わせることで、より強固な管理体制となります。
試験機のメンテナンスとメーカーサポートの重要性
試験機自体も経年劣化します。特に、圧力を感知するセンサー部分や、ポートを密閉するパッキン(シール材)は消耗品です。
パッキンが劣化すると、グローブは正常なのに「リークあり」と誤判定される原因になります。
定期的な消耗品交換や、年に1回のメーカーによる校正・点検を計画に組み込みましょう。また、トラブル発生時に迅速に対応してくれる国内サポート体制が整っているメーカーを選ぶことも、長期的な安定稼働の鍵となります。
まとめ

グローブリーク試験は、再生医療等の製造現場において、無菌性を保証するための「最後の砦」を守る極めて重要なプロセスです。
PIC/S GMP Annex 1の改訂により、その管理基準はますます厳格化しており、科学的根拠に基づいた試験の実施とデータ管理が求められています。
適切な試験機の選定、バリデーションされた試験条件の設定、そして日常的な運用管理を徹底することで、ピンホールによる汚染リスクを最小限に抑えることができます。
現場の負担を考慮しつつ、確実な品質保証体制を構築することは、製品の価値を高め、患者様の信頼に応えることにつながるでしょう。ぜひ、自社の運用に最適なグローブ管理を見直してみてください。
グローブリーク試験についてよくある質問
現場のご担当者様からよく寄せられる質問をまとめました。運用のヒントとしてご活用ください。
- Q1. グローブリーク試験の頻度はどのくらいが適切ですか?
- 基本的には、製造作業の「前」と「後」の両方で実施することが推奨されています。特にPIC/S GMP Annex 1ではこの運用が求められています。ただし、連続生産(キャンペーン生産)の場合は、リスク評価に基づいて適切な間隔を設定することも可能です。
- Q2. どの程度の大きさのピンホールまで検知できますか?
- 一般的なグローブリークテスターでは、100μm(マイクロメートル)程度のピンホール検知を基準とすることが多いです。機種や設定条件によっては、より微細な穴の検知も可能ですが、感度を上げすぎると測定時間が長くなったり、環境ノイズの影響を受けやすくなったりするため、バランスが重要です。
- Q3. 目視検査だけでは不十分なのでしょうか?
- はい、不十分です。100μmレベルの微細な穴は肉眼では発見困難です。また、目視検査は作業者のスキルや体調に左右されやすく、客観的な記録も残りません。物理的な試験機による定量的な検査との併用が必須です。
- Q4. 試験に時間がかかりすぎて生産効率が落ちています。対策はありますか?
- 複数のポートを同時に測定できる多連式のシステムや、高速測定アルゴリズムを搭載した最新機種への更新を検討してみてください。また、ワイヤレス機を複数台導入して並行作業を行うことでも、トータルの準備時間を短縮できます。
- Q5. グローブの材質によって試験条件は変わりますか?
- はい、変わります。CSM、ネオプレン、EPDMなど、材質によって伸縮性やガス透過性が異なるため、圧力設定や安定化時間を調整する必要があります。導入時のバリデーション(PQ)で、材質ごとに最適なパラメータを設定しましょう。